

Potential biomarkers of resistance to CDK4/6 inhibitors: a narrative review of preclinical and clinical studies
Introduction
Breast cancer (BC) is the most common cancer diagnosed in women with the morbidity ranging from 27/100,000 to 94/100,000 (1), which accounts for 25% of all malignancies worldwide (2). BC is generally categorized into 3 subtypes: hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative BC (HR+/HER2– BC), HER2–positive (HER2+) BC and triple negative BC (3). Of all BC cases, approximately 60–70% of women are diagnosed as HR+/HER2– BC (4). In the past decades, the standard of care for HR+/HER2– BC is blocking the estrogen receptor signaling pathway, which is so-called endocrine therapy (ET) (5). Although the overall 5-year survival has increased to 90%, remains incurable with an estimated 5-year survival rate was only 25% in metastatic or advanced BC (mBC/ABC) (6). Thus, new therapeutic regimens are continuously explored to extend the overall survival (OS) of BC patients.
With the discovery of cyclin-dependent kinase (CDK) 4/6 inhibitors, the progression-free survival (PFS) is almost doubled when combination of CDK4/6 inhibitors and aromatase inhibitors (AIs) as the first line treatment of HR+/HER2– mBC/ABC patients (Table 1) (7-9). Even in patients progressed with previously ET, the PFS is significantly prolonged in combination with fulvestrant (10-12). In addition, the statistically significant OS data were also reported in MONALEESA-3 (13), MONALEESA-7 (14) and MONARCH 2 (Table 1) (15). These promising results make the combination of CDK4/6 inhibitors and ET as a standard of care for treatment of HR+/HER2– mBC/ABC, which is highly recommended in various BC guidelines (16,17).
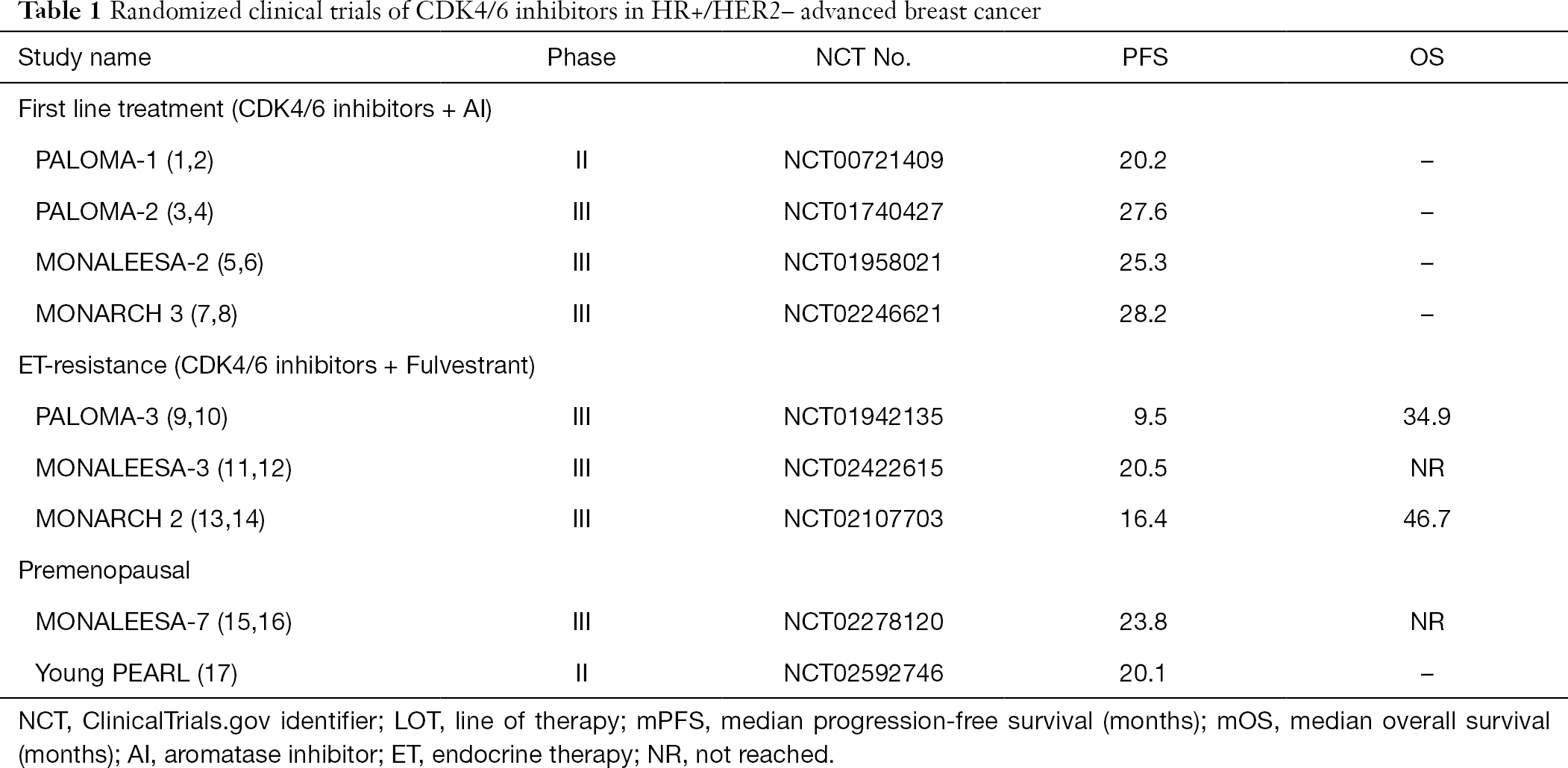
Full table
However, not all HR+/HER2– BC patients achieved clinical benefits from CDK4/6 inhibitor-based therapy and almost all the patient would gained the acquired resistance finally (18). The mechanisms of resistance to CDK4/6 inhibitors is still unclear and there were no predictive biomarkers indicating the clinical efficacy of CDK4/6 inhibitors. This review mainly discussed the pharmacological mechanism of CDK4/6 inhibitors and the potential biomarkers of resistance to CDK4/6 inhibitors (Table 2), aiming to provide some insights to overcome the resistance to CDK4/6 inhibitors. We present the following review in accordance with the Narrative Review reporting checklist (available at http://dx.doi.org/10.21037/tbcr-20-52).

Full table
Pharmacological mechanism of CDK4/6 inhibitors
In normal physiological conditions, cell cycle is tightly controlled to maintain the normal process of cell proliferation. Dysregulation of cell cycle involved in many pathological process, including cancers (19). Cell cycle is classically divided into four phases: G1 (pre-DNA synthesis), S (DNA synthesis), G2 (pre-division), and M (cell division) (20). Many signaling pathways have involved into cell cycle regulation and CDK4/6 is one of the most important kinase initiating cell cycle transition from G1 phase to S phase (21). In G1 phase of cell cycle, activation of upstream signaling pathways promotes the combination of CDK4/6 with cyclin D (Figure 1) (22). The formation of cyclin D-CDK4/6 complex released adenosine triphosphate (ATP), which provides energy for phosphorylation of retinoblastoma (Rb) (23,24). Inactivated Rb is tightly binding to E2 transcription factor (E2F) to repress the E2F function (25). When phosphorylated, Rb releases E2F from Rb-E2F complex and then the dissociated E2F induced the upregulation of target genes and initiating DNA replication, resulting in cell cycle transition from G1 phase to S phase (20,26).

CDK4 and CDK6 are expressed in most cell types and share 71% amino acid identity (27). The functions of CDK4 and CDK6 are largely overlapped and both of them can partner with all 3 D-type cyclins (D1, D2 and D3) (27). Intrinsically, CDK4/6 activity is inhibited by the INK4 family (p16, p15, p18, and p19) and by the Cip (p21) and Kip (p27) family (28). In BC, cyclin D-CDK4/6-Rb signaling cascade was dysregulated, which accelerated the unchecked cell proliferation (29,30). Amplification of cyclin D1 gene was observed in ~15% of BC cases and cyclin D1 mRNA and protein was overexpressed in up to 50% of primary breast cancers, mostly ER-positive and well-differentiated tumors (31,32). In breast cancer cell lines, upregulation of cyclin D induced cell cycle progressing from G1 to S phase (33). In transgenic mice, excessive expression of cyclin D accelerated cell proliferation, contributing to the progression of mammary carcinomas (34). Cyclin D is often overexpressed in many cancers and correlated with poor prognosis and high metastasis of tumor (31,35,36). Similarly, upregulation of CDK4 expression is positively correlated with high tumor cell proliferation in sporadic breast carcinomas (37). Suppression of cyclin D3-CDK6 signaling resulted in tumor cell apoptosis (38). Therefore, both CDK4/6 and cyclin D play a crucial role in caner progression and could be the therapeutic targets for cancer treatment.
So far, there are three CDK4/6 inhibitors approved by U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA): palbociclib (Ibrance, Pfizer, USA), ribociclib (Kisqali, Novartis, Switzerland) and abemaciclib (Verzenio, Lilly, USA). The indications of these 3 CDK4/6 inhibitors approved by FDA and EMA are: (I) in combination with an AI as initial therapy in postmenopausal women with HR+/HER2– ABC/mBC; (II) in combination with fulvestrant for women who have previously treated with ET (39). Moreover, abemaciclib is the only CDK4/6 inhibitor that can be used as a monotherapeutic drug and palbociclib is the only one which is approved to treat male patients with HR+/HER2– mBC on the basis of real-world data on April 4, 2019 by FDA (40). In China, only palbociclib has been approved by National Medical Products Administration (NMPA) in 2017 and is orally administered in combination with AI as initial therapy in postmenopausal women with HR+/HER2– ABC/mBC.
Potential biomarkers of resistance to CDK4/6 inhibitors
Resistance to CDK4/6 inhibitors: regulation of cell cycle (Table 2)
p16 amplification
p16INK4A, encoded by the CDKN2Aink4a, is an intrinsic tumor-suppressor which can bind to CDK4/6 to block the interaction between CDK4/6 and cyclin D, resulting in the inactivation of the downstream signaling pathway (Figure 1) (41-43). The expression of p16 is upregulated during oncogenic stress. When p16 amplification is concurrent with Rb mutation, resistance to CDK4/6 inhibitor was observed due to loss of Rb function (44). In the presence of Rb, overexpression of p16 demonstrated resistance to CDK4/6 inhibitor through downregulation of CDK4 in breast cancer cells (18,45). In melanoma cell lines, downregulation of CDKN2Aink4a expression is associated with the sensitivity to palbociclib (46). Deletion of p16 demonstrated a significant repression of cell cycle in G1 phase and inhibited cell proliferation by palbociclib in glioblastoma xenograft cells (47). All these preclinical evidences indicated that p16 amplification should be an effective biomarker of resistance to CDK4/6 inhibitors. However, the data of PALOMA-1 study did not present any significant difference in PFS between the unselected cohort and the loss of p16/CCND1 amplification cohort (48). Similar results were also observed from the biomarker analysis of PALOMA-2 and PALOMA-3 (49,50). Therefore, it is controversial to use p16 amplification as a biomarker.
CCND1 amplification
There are 3 isoforms of D cyclins, cyclins D1, D2 and D3, all of which could form a complex with CDK4/6, driving cell cycle transition from G1 to S phase (51). Cyclins could allosterically activate the cognate CDKs through formation of cyclin-CDK complexes, which contribute to cell cycle progression (32,52). Overexpression of D cyclins can promote cell proliferation and induce tumor growth (32). Among these 3 D cyclins, cyclin D1 is more frequently overexpressed than cyclin D2 and D3 in human cancers (52). The cyclin D1 encoding gene, CCND1 have been found to upregulated in ER+ BC (cyclin D1; 58% in luminal B vs. 29% in luminal A) (53). Therefore, CCND1 amplification or cyclin D1 overexpression would be an indicator of resistance of CDK4/6 inhibitor. Unfortunately, the biomarker analysis of PALOMA studies did not show any significant difference on PFS according the expression level of CCND1 (49,50,54). The role of CCND1 in CDK4/6 inhibition should be further explored.
CDK4 or CDK6 amplification
Both CDK4 and CDK6 are the major components of cyclin D-CDK4/6-Rb signaling pathway (55). As mentioned above, CDK4 and CDK6 are expressed in most cell types and share 71% amino acid identity and their functions are largely overlapped (27). Previous studies have revealed that the cell proliferation of mouse embryonic fibroblast was normal but have a delayed S phase in the CDK4 null mouse model (56). In CDK4-/- mice, the expression of CDK6 was upregulated in lung (57). Thus, lack of CDK4 function might be compensated by increased level of CDK6 in these models. On the basis of these observations, CDK6 seems to be more important than CDK4. In CDK4-amplified glioblastoma cells, pRb is persistent even if the cells were treated with palbociclib (47). Reduction of sensitivity to ribociclib was also observed in fusion-positive rhabdomyosarcoma with CDK4 amplification and overexpression (58). These preclinical data suggested that CDK4 amplification might indicate a poor response to CDK4/6 inhibitors. In HR+ breast cancer cells, prolonged exposure to abemaciclib induced amplification of CDK6, which reduced pRb and sensitivity to all the three CDK4/6 inhibitors, demonstrating that CDK6 amplification could be the mechanism of acquired resistance to CDK4/6 inhibitors (59). However, biomarker analysis of PALOMA-2 and PALOMA-3 did not support these preclinical findings that amplification of CDK4 or CDK6 was a biomarker to predict the prognosis of mBC/ABC patients treated with CDK4/6 inhibitors (49,50). But the data of ECLIPS study showed that high baseline CDK4 expression indicated a longer PFS compared with patients who had a low baseline CDK4 expression (low vs. high: 6.45 months vs. not reached, P=0.01) in patients treated with palbociclib plus fulvestrant (60). Therefore, the predicted value of CDK4 or CDK6 amplification in early BC should be investigated in future.
CCNE1-CDK2 amplification
In addition to cyclin D-CDK4/6 complex, cyclin E-CDK2 complex could also release E2F via phosphorylating Rb (61). Cyclin E is expressed in the late G1 phase until the end of S phase of cell cycle (62). It can bind and activate the kinase CDK2 to control the entry of S phase (63). Activation of cyclin E-CDK2 complex phosphorylates Rb and activate E2F to upregulate the target gene expression, which is required for S phase (64). Upregulation of cyclin E expression was associated with tumorigenesis of human cancers, especially BC (62). CCNE1 expression was amplified in CDK4/6 inhibitor resistance cell lines (65) and upregulation of Cyclin E1 indicated the reduction of the inhibitory effects of CDK4/6 inhibitors on cell cycle progression (66). Biomarker analysis of PALOMA-3 demonstrated that overexpression of CCNE1 mRNA is associated with a shorter PFS in patients treated with the combination of palbociclib and fulvestrant (50) while this was not found in PALOMA-2 study (49), suggesting that upregulation of cyclinE1 mRNA was one of the mechanisms of resistance to CDK4/6 inhibitors in previously treated HR+/HER2– mBC.
CDK7 amplification
Unlike CDK4/6, CDK7 has downstream effects as transcriptional regulators and cell cycle regulator through formation of complex with cyclin H or MAT1 (67,68). Evidence has been revealed that CDK7-cyclin H complex also functioned as CDK activating kinase (CAK) (69), which can activate cyclin T1-CDK9 and cyclin K-CDK12/13 resulting in a pause of transcription (70). Furthermore, as a CAK, CDK7 can activate CDK4/6 to regulate G1 phase transition, maintain CDK1 and CDK 2 activity to involve into G2/M phase (68). After treated with palbociclib, the expression of CDK7 was increased by 27.27% in hematopoietic stem cells in mice (71). Upregulation of CDK7 demonstrated the resistance to palbociclib in MCF7 cells (72). Thus, overexpression of CDK7 might be a biomarker of acquired resistance to CDK4/6 inhibitors. However, neither PALOMA-2 nor PALOMA-3 results analyzed the change of CDK7 expression as a biomarker of CDK4/6 inhibitors. More clinical evidence is required to verify the role of CDK7 in resistance to CDK4/6 inhibitors.
CDK9 amplification
Similar to CDK7, CDK9 also has downstream effects as transcriptional regulators (67), which is crucial to RNA Polymerase II (Pol II) transcription initiation, elongation, and termination (73). Activation of CDKs is mainly dependent on the formation of heterodimers with cyclin proteins (74). CDK9 interacts with cyclin T to form the Positive Transcription Elongation Factor b (P-TEFb) (75,76), resulting in activating the transcription elongation of the target genes in nucleus, including MYC, MCL1 and NF-κB (77,78). CDK9 is constitutively expressed throughout cell cycle and overexpressed in many cancers, such as lung cancer, ovarian cancer, leukemia, prostate cancer, as well as breast cancer (79,80). Results from ECLIPS study have shown that the increased number of copies/mL of CDK9 in plasma-derived exosomes was observed after 3 months of treatment compared with before treatment (3,800 vs. 7,500 copies/mL, P=0.03) in HR+/HER2– mBC patients whose disease has progressed after treated with palbociclib plus ET (60), indicating that overexpression of CDK9 is positively correlated with clinical resistance of CDK4/6 inhibitors.
Loss of Rb function
Rb, as the downstream protein of CDK4/6, is considered as the most important biomarker indicating the clinical activity to CDK4/6 inhibitors (81). Preclinical studies have proved that loss of Rb function was detected in palbociclib resistance cell lines (82). A phase II RCT study, POP study, aimed to investigate the effects of palbociclib on cell proliferation in the neoadjuvant setting in BC patients (83). The results manifested that early decreased of Rb phosphorylation (pRb) was associated with antiproliferative response to palbociclib, suggesting that pRb would be a potential indicator to identify patients with primary resistance. Somatic Rb1 mutations were found when disease was progressed in mBC patients who had received the treatment with palbociclib or ribociclib (84), suggesting that Rb mutation might be an effective indicator to the acquired resistance to CDK4/6 inhibitors. However, there were only 3 patients in this publication. More patients and real-world data were warranted to confirm this conclusion.
E2F amplification
As mentioned above, CDK-Rb-E2F axis is critical for driving cell cycle progression. Expression and activity of E2E is strictly controlled in the normal physiological process at different levels, including transcription, mRNA stability, post-translational modifications, interaction with regulatory proteins and protein stability (85). Upregulation of E2F and E2F target in cancer is associated with poor prognosis (85). The dissociation of Rb-E2F complex is regulated by cyclin D-CDK4/6 and cyclin E-CDK2 (86). In turn, cyclin D3 and cyclin E are the target gene of E2F (87-89). As the downstream signaling, activation of E2F by the bypass of cyclin D-CDK4/6, such as cyclin E-CDK2, might be the mechanism of resistance to CDK4/6 inhibitors.
Resistance to CDK4/6 inhibitors: regulation beyond cell cycle (Table 2)
TK1
Thymidine kinase-1 (TK1) is a key regulator of cell cycle, which is highly expressed in S/G2 phase, and functioned as an enzyme to catalyze the synthesis of DNA precursor (90). Serum TK1 level and activity was significantly increased in solid tumors, including breast cancer, lung and colorectal (91). In primary BC patients, increased TK1 levels and activity is positively correlated with large tumor size and poor outcomes (92,93). In HR+/HER2– mBC patients, lower baseline TK1 activity was associated with a longer PFS and reduction of TK1 activity after one month of treatment was also associated a significantly prolongation of PFS (94). Thus, TK1 might be a meaningful prognostic biomarker for HR+/HER2– mBC. The prospective, pharmacogenetic study, ECLIPS, aims to explore the predictive biomarkers of responsive/resistant to the combined treatment of palbociclib and ET (letrozole or fulvestrant) (60). The data demonstrated that the number of copies/mL of TK1 was significantly increased after 3 months of treatment compared with before treatment (1,200 vs. 3,350 copies/mL, P=0.01) in disease progressed patients. In addition, results of TREnd trial showed that increased level of TK1 mRNA is linked to shorter PFS (95). These data supported that TK1 level might be a significant biomarker of acquired resistance to CDK4/6 inhibitors.
Loss of FAT1
FAT1 is a tumor-suppressor belonging to the cadherin superfamily and interacts with the Wnt/β-catenin and Hippo signaling pathways (96-98). Loss of FAT1 function contributed to the development and progression of cancers (96). Gene sequencing results manifested that FAT1 mutation accounted for ~2% in primary and ~6% in metastatic tumors in HR+/HER2– BC patients (99). Preclinical data demonstrated that loss of FAT1 resulted in excessive expression of CDK6 through activation of Hippo pathway, contributing to resistance to CDK4/6 inhibitors (100). Gene analysis results from 348 ER+/HER2– BC patients previously treated with CDK4/6 inhibitors, manifested that loss of FAT1 was linked with poor prognosis of CDK4/6 inhibitor-based therapy, with a shorter PFS (2.4 months) than that in FAT1 wild type arm (PFS: 10.1 months; P=2.2×10–11) (100). Therefore, loss of FAT1 might be an effective biomarker of resistance to CDK4/6 inhibitors.
Activation of the PI3K/Akt/mTOR pathway
Phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) signaling pathway is activated in ~30–40% of BC, especially in HR+ BC (65,101). PI3K interacts with phosphorylated tyrosines on IRS molecules, which further phosphorylates and activates Akt (102). Phosphorylation of Akt can activate mTOR, a serine/threonine kinase laying downstream of the PI3K/Akt/mTOR pathway, leading to the upregulation of mRNA translation, protein synthesis, as well as cell proliferation (103). Kinome-wide siRNA screen results has revealed that Akt pathway was excessively activated via phosphorylation of Ak at S477/T479 in ribociclib-resistance breast cancer cells and that the level PDK1, a downstream kinase of Akt was increased after short- and long-term treatment with ribociclib in MCF-7 cells, indication that Akt-PDK1 pathway mediated acquired resistance to ribociclib (104). Dual blockade of mTOR and CDK4/6 has a synergistically inhibitory effects on E2F-dependent transcription (105). In a PDX model, the acquired resistance to ribociclib was attenuated by combination of ribociclib and alpelisib. Triple combination of fulvestrant, ribociclib and alpelisib was a better effect on repressing rapid tumor progression that paired combinations (65). All these results indicated that inhibition of PI3K/Akt/mTOR signaling pathway may overcome the acquired resistance to CDK4/6 inhibitors.
FGFR amplification
Fibroblast growth factor receptor (FGFR) is a type of tyrosine kinase receptors (106). Activation of FGFR signaling can transduce the activation of PI3K/Akt/mTOR pathway, MAPK/ERK pathway, STAT3 pathway, as well as ribosomal protein S6 kinase 2, leading to cell survival, proliferation, differentiation, etc. (106). Overexpression of FGFR was associated with poor prognosis with the reduction of OS and also involved in treatment resistance in various cancers, including breast cancer (107,108). Palbociclib resistant cells showed the increased activity of EKR1/2 and mTOR and overexpression of CDK6, cyclin D and cyclin E, which was driven by FGFR1 signaling in non-small cell lung cancer, indicating that FGFR1-MAP kinase-mTOR pathway contributed to palbociclib resistance through upregulation of CDK6 and cyclin D expression (109). In ER+ BC cells, FGFR1 amplification demonstrated the resistance to fulvestrant ± ribociclib/palbociclib. FGFR1/2 amplification or activating mutations were identified in 14 of 34 patients who have progressed after treatment with CDK4/6 inhibitors by next generation sequencing (NGS) of circulating tumor DNA (ctDNA) (110). Furthermore, data of MONALESSA-2 trial (ribociclib plus letrozole arm) showed that patients with FGFR1 amplification in ctDNA has a shorter PFS (10.61 months) than that (24.84 months) in patients without FGFR1 amplification (110). Taken together, amplification of FGFR1 should be considered as a useful biomarker of resistance to CDK4/6 inhibitors.
Immune regulation
The promising data and clinical success of immune checkpoint inhibition is the milestone of cancer immunotherapy. Results of KEYNOTE-522 showed that the pathological complete response rate increased to 64.8% with combination of pembrolizumab and neoadjuvant chemotherapy compared with 51.2% in the neoadjuvant chemotherapy in patients with early triple negative BC, demonstrating the potential clinical use of pembrolizumab in BC (111). CDK4/6 and cyclin D have been revealed to play a role in immune cells (112). Cyclin D3 is required for early development of B cells by integrating cytokine and is essential for maturation of T lymphocyte (113,114). Inhibition of CDK4/6 may promote the infiltration of immune cells through senescence-associated secretory phenotype (115). Inhibition of CDK4/6 by palbociclib or trilaciclib increased T cell activation but decreased T cell proliferation. Combination of CDK4/6 inhibitors and anti-PD-1 antibody showed a synergistic effects on anti-tumor immunity, which is mediated by enhancement of T cell activation (116). These preclinical studies suggest that immune regulation may influence the anti-tumor activity of CDK4/6 inhibitors. Interestingly, biomarker analysis of PALOMA-2 found that higher level of PD-1 showed less clinical benefit from the combination of palbociclib and letrozole compared with low PD-1expression (49). Taken together, immune regulation may also one of the mechanisms of resistance to CDK4/6 inhibitors.
Conclusions
In conclusion, this review summarized the mechanism of CDK4/6 inhibitors, preclinical and potential clinical biomarkers of resistance to CDK4/6 inhibitors. Resistance to CDK4/6 inhibitor was mainly caused by upregulation of downstream proteins, excessive activation of downstream signaling pathways, and activation of bypass signaling pathways. However, most of the biomarkers mentioned in this review are under preclinical research. Only few of them have been validated by clinical data. Therefore, large clinical trials and real-world evidences are warranted to figure out the resistant mechanism of CDK4/6 inhibitors, which will facilitate oncologists to maximize clinical benefits of CDK4/6 inhibitors and optimize the therapeutic regimens after disease progression.
Acknowledgements
The authors would like to acknowledge Pfizer Medical Affairs (China) for their scientific comments on this manuscript.
Funding: None.
Footnote
Reporting Checklist: The authors have completed the Narrative Review reporting checklist. Available at http://dx.doi.org/10.21037/tbcr-20-52
Conflicts of Interest: Both authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/tbcr-20-52). The authors have no conflicts of interest to declare.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
References
- Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur J Cancer 2013;49:1374-403. [Crossref] [PubMed]
- Ghoncheh M, Pournamdar Z, Salehiniya H. Incidence and Mortality and Epidemiology of Breast Cancer in the World. Asian Pac J Cancer Prev 2016;17:43-6. [Crossref] [PubMed]
- Lettieri CK, Garcia-Filion P, Hingorani P. Incidence and outcomes of desmoplastic small round cell tumor: results from the surveillance, epidemiology, and end results database. J Cancer Epidemiol 2014;2014:680126 [Crossref] [PubMed]
- Serra F, Lapidari P, Quaquarini E, et al. Palbociclib in metastatic breast cancer: current evidence and real-life data. Drugs Context 2019;8:212579 [Crossref] [PubMed]
- Fernandes MT, Adashek JJ, Barreto CMN, et al. A paradigm shift for the treatment of hormone receptor-positive, human epidermal growth factor receptor 2-negative (HR+/HER2-) advanced breast cancer: a review of CDK inhibitors. Drugs Context 2018;7:212555 [Crossref] [PubMed]
- Institute NC. Cancer Stat Facts: Female Breast Cancer. 2019. Available online: https://seer.cancer.gov/statfacts/html/breast.html
- Finn RS, Martin M, Rugo HS, et al. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med 2016;375:1925-36. [Crossref] [PubMed]
- Hortobagyi GN, Stemmer SM, Burris HA, et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N Engl J Med 2016;375:1738-48. [Crossref] [PubMed]
- Goetz MP, Toi M, Campone M, et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J Clin Oncol 2017;35:3638-46. [Crossref] [PubMed]
- Sledge GW Jr, Toi M, Neven P, et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2- Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J Clin Oncol 2017;35:2875-84. [Crossref] [PubMed]
- Turner Nicholas C, Ro Jungsil, André Fabrice, et al. Palbociclib in Hormone-Receptor–Positive Advanced Breast Cancer. N Engl J Med 2015;373:1672-3. [Crossref] [PubMed]
- Slamon DJ, Neven P, Chia S, et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer: MONALEESA-3. J Clin Oncol 2018;36:2465-72. [Crossref] [PubMed]
- Slamon DJ, Neven P, Chia S, et al. Overall Survival with Ribociclib plus Fulvestrant in Advanced Breast Cancer. N Engl J Med 2020;382:514-24. [Crossref] [PubMed]
- Im SA, Lu YS, Bardia A, et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N Engl J Med 2019;381:307-16. [Crossref] [PubMed]
- Sledge GW Jr, Toi M, Neven P, et al. The Effect of Abemaciclib Plus Fulvestrant on Overall Survival in Hormone Receptor-Positive, ERBB2-Negative Breast Cancer That Progressed on Endocrine Therapy-MONARCH 2: A Randomized Clinical Trial. JAMA Oncol 2020;6:116-24. [Crossref] [PubMed]
- Gradishar WJ, Anderson BO, Abraham J, et al. Breast Cancer, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2020;18:452-78. [Crossref] [PubMed]
- Xu F, Jiang Z. CSCO BC guideline: updates for hormone receptor-positive breast cancer in 2020. Transl Breast Cancer Res 2020;1:3. [Crossref]
- Pandey K, An HJ, Kim SK, et al. Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: A review. Int J Cancer 2019;145:1179-88. [Crossref] [PubMed]
- Ingham M, Schwartz GK. Cell-Cycle Therapeutics Come of Age. J Clin Oncol 2017;35:2949-59. [Crossref] [PubMed]
- Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev 2016;45:129-38. [Crossref] [PubMed]
- Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, et al. Targeting cell cycle regulation in cancer therapy. Pharmacol Ther 2013;138:255-71. [Crossref] [PubMed]
- O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 2016;13:417-30. [Crossref] [PubMed]
- Narasimha AM, Kaulich M, Shapiro GS, et al. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife 2014;3:e02872 [Crossref] [PubMed]
- Harbour JW, Luo RX, Santi AD, et al. Cdk Phosphorylation Triggers Sequential Intramolecular Interactions that Progressively Block Rb Functions as Cells Move through G1. Cell 1999;98:859-69. [Crossref] [PubMed]
- Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev 2000;14:2393-409. [Crossref] [PubMed]
- Stevaux O, Dyson NJ. A revised picture of the E2F transcriptional network and RB function. Curr Opin Cell Biol 2002;14:684-91. [Crossref] [PubMed]
- Grossel MJ, Hinds PW. From cell cycle to differentiation: an expanding role for cdk6. Cell Cycle 2006;5:266-70. [Crossref] [PubMed]
- Baker SJ, Reddy EP. CDK4: A Key Player in the Cell Cycle, Development, and Cancer. Genes Cancer 2012;3:658-69. [Crossref] [PubMed]
- Mayer EL. Targeting breast cancer with CDK inhibitors. Curr Oncol Rep 2015;17:443. [Crossref] [PubMed]
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009;9:153-66. [Crossref] [PubMed]
- Taneja P, Maglic D, Kai F, et al. Classical and Novel Prognostic Markers for Breast Cancer and their Clinical Significance. Clin Med Insights Oncol 2010;4:15-34. [Crossref] [PubMed]
- Musgrove EA, Caldon CE, Barraclough J, et al. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 2011;11:558-72. [Crossref] [PubMed]
- Musgrove EA, Lee CS, Buckley MF, et al. Cyclin D1 induction in breast cancer cells shortens G1 and is sufficient for cells arrested in G1 to complete the cell cycle. Proc Natl Acad Sci U S A 1994;91:8022-6. [Crossref] [PubMed]
- Wang TC, Cardiff RD, Zukerberg L, et al. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature 1994;369:669-71. [Crossref] [PubMed]
- Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer 2007;7:750-62. [Crossref] [PubMed]
- Roy PG, Thompson AM. Cyclin D1 and breast cancer. Breast 2006;15:718-27. [Crossref] [PubMed]
- An HX, Beckmann MW, Reifenberger G, et al. Gene amplification and overexpression of CDK4 in sporadic breast carcinomas is associated with high tumor cell proliferation. Am J Pathol 1999;154:113-8. [Crossref] [PubMed]
- Wang H, Nicolay BN, Chick JM, et al. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 2017;546:426-30. [Crossref] [PubMed]
- Wu Y, Zhang Y, Pi H, et al. Current Therapeutic Progress of CDK4/6 Inhibitors in Breast Cancer. Cancer Manag Res 2020;12:3477-87. [Crossref] [PubMed]
- Wedam S, Fashoyin-Aje L, Bloomquist E, et al. FDA Approval Summary: Palbociclib for Male Patients with Metastatic Breast Cancer. Clin Cancer Res 2020;26:1208-12. [Crossref] [PubMed]
- Siebert R, Willers CP, Opalka B. Role of the cyclin-dependent kinase 4 and 6 inhibitor gene family p15, p16, p18 and p19 in leukemia and lymphoma. Leuk Lymphoma 1996;23:505-20. [Crossref] [PubMed]
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993;366:704-7. [Crossref] [PubMed]
- Russo AA, Tong L, Lee JO, et al. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 1998;395:237-43. [Crossref] [PubMed]
- Liu Y, Zhong X, Wan S, et al. p16(INK4a) expression in retinoblastoma: a marker of differentiation grade. Diagn Pathol 2014;9:180. [Crossref] [PubMed]
- Dean JL, McClendon AK, Hickey TE, et al. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 2012;11:2756-61. [Crossref] [PubMed]
- Young RJ, Waldeck K, Martin C, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res 2014;27:590-600. [Crossref] [PubMed]
- Cen L, Carlson BL, Schroeder MA, et al. p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells. Neuro Oncol 2012;14:870-81. [Crossref] [PubMed]
- Finn RS, Crown JP, Ettl J, et al. Efficacy and safety of palbociclib in combination with letrozole as first-line treatment of ER-positive, HER2-negative, advanced breast cancer: expanded analyses of subgroups from the randomized pivotal trial PALOMA-1/TRIO-18. Breast Cancer Res 2016;18:67. [Crossref] [PubMed]
- Finn RS, Liu Y, Zhu Z, et al. Biomarker Analyses of Response to Cyclin-Dependent Kinase 4/6 Inhibition and Endocrine Therapy in Women with Treatment-Naïve Metastatic Breast Cancer. Clin Cancer Res 2020;26:110-21. [Crossref] [PubMed]
- Turner NC, Liu Y, Zhu Z, et al. Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor–Positive Metastatic Breast Cancer. J Clin Oncol 2019;37:1169-78. [Crossref] [PubMed]
- Kato JY, Sherr CJ. Inhibition of granulocyte differentiation by G1 cyclins D2 and D3 but not D1. Proc Natl Acad Sci U S A 1993;90:11513-7. [Crossref] [PubMed]
- Qie S, Diehl JA. Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med (Berl) 2016;94:1313-26. [Crossref] [PubMed]
- Araki K, Miyoshi Y. Mechanism of resistance to endocrine therapy in breast cancer: the important role of PI3K/Akt/mTOR in estrogen receptor-positive, HER2-negative breast cancer. Breast Cancer 2018;25:392-401. [Crossref] [PubMed]
- Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 2015;16:25-35. [Crossref] [PubMed]
- Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov 2016;6:353-67. [Crossref] [PubMed]
- Rane SG, Dubus P, Mettus RV, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet 1999;22:44-52. [Crossref] [PubMed]
- Tsutsui T, Hesabi B, Moons DS, et al. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol 1999;19:7011-9. [Crossref] [PubMed]
- Olanich ME, Sun W, Hewitt SM, et al. CDK4 Amplification Reduces Sensitivity to CDK4/6 Inhibition in Fusion-Positive Rhabdomyosarcoma. Clin Cancer Res 2015;21:4947-59. [Crossref] [PubMed]
- Yang C, Li Z, Bhatt T, et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017;36:2255-64. [Crossref] [PubMed]
- Del Re M, Bertolini I, Crucitta S, et al. Overexpression of TK1 and CDK9 in plasma-derived exosomes is associated with clinical resistance to CDK4/6 inhibitors in metastatic breast cancer patients. Breast Cancer Res Treat 2019;178:57-62. [Crossref] [PubMed]
- Obeyesekere MN, Herbert JR, Zimmerman SO. A model of the G1 phase of the cell cycle incorporating cyclin E/cdk2 complex and retinoblastoma protein. Oncogene 1995;11:1199-205. [PubMed]
- Möröy T, Geisen C, Cyclin E. Int J Biochem Cell Biol 2004;36:1424-39. [Crossref] [PubMed]
- Teixeira LK, Reed SI, Cyclin E. Deregulation and Genomic Instability. Adv Exp Med Biol 2017;1042:527-47. [Crossref] [PubMed]
- Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer 2002;2:910-7. [Crossref] [PubMed]
- Herrera-Abreu MT, Palafox M, Asghar U, et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res 2016;76:2301-13. [Crossref] [PubMed]
- Taylor-Harding B, Aspuria PJ, Agadjanian H, et al. Cyclin E1 and RTK/RAS signaling drive CDK inhibitor resistance via activation of E2F and ETS. Oncotarget 2015;6:696-714. [Crossref] [PubMed]
- Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci 2005;30:630-41. [Crossref] [PubMed]
- Schachter MM, Merrick KA, Larochelle S, et al. A Cdk7-Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol Cell 2013;50:250-60. [Crossref] [PubMed]
- Harper JW, Elledge SJ. The role of Cdk7 in CAK function, a retro-retrospective. Genes Dev 1998;12:285-9. [Crossref] [PubMed]
- Fisher RP. Cdk7: a kinase at the core of transcription and in the crosshairs of cancer drug discovery. Transcription 2019;10:47-56. [Crossref] [PubMed]
- Ji Q, Qi R, Zhang L, et al. Effect of PD0332991 on biological activity of hematopoietic stem cells in mice. Zhonghua Xue Ye Xue Za Zhi 2014;35:157-61. [PubMed]
- Martin LA, Pancholi S, Ribas R, et al. Abstract P3-03-09: Resistance to palbociclib depends on multiple targetable mechanisms highlighting the potential of drug holidays and drug switching to improve therapeutic outcome. Cancer Res 2017;77:P3-03-9.
- Bacon CW, D'Orso I. CDK9: a signaling hub for transcriptional control. Transcription 2019;10:57-75. [Crossref] [PubMed]
- Hochegger H, Takeda S, Hunt T. Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat Rev Mol Cell Biol 2008;9:910-6. [Crossref] [PubMed]
- Wei P, Garber ME, Fang SM, et al. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998;92:451-62. [Crossref] [PubMed]
- Peng J, Marshall NF, Price DH. Identification of a cyclin subunit required for the function of Drosophila P-TEFb. J Biol Chem 1998;273:13855-60. [Crossref] [PubMed]
- Taube R, Lin X, Irwin D, et al. Interaction between P-TEFb and the C-terminal domain of RNA polymerase II activates transcriptional elongation from sites upstream or downstream of target genes. Mol Cell Biol 2002;22:321-31. [Crossref] [PubMed]
- Zhang H, Pandey S, Travers M, et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018;175:1244-58.e26. [Crossref] [PubMed]
- Wang J, Dean DC, Hornicek FJ, et al. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in ovarian cancer. FASEB J 2019;33:5990-6000. [Crossref] [PubMed]
- Garriga J, Bhattacharya S, Calbó J, et al. CDK9 is constitutively expressed throughout the cell cycle, and its steady-state expression is independent of SKP2. Mol Cell Biol 2003;23:5165-73. [Crossref] [PubMed]
- Wiedemeyer WR, Dunn IF, Quayle SN, et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc Natl Acad Sci U S A 2010;107:11501-6. [Crossref] [PubMed]
- Malorni L, Piazza S, Ciani Y, et al. A gene expression signature of retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget 2016;7:68012-22. [Crossref] [PubMed]
- Arnedos M, Bayar MA, Cheaib B, et al. Modulation of Rb phosphorylation and antiproliferative response to palbociclib: the preoperative-palbociclib (POP) randomized clinical trial. Ann Oncol 2018;29:1755-62. [Crossref] [PubMed]
- Condorelli R, Spring L, O’Shaughnessy J, et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol 2018;29:640-5. [Crossref] [PubMed]
- Kent LN, Leone G. The broken cycle: E2F dysfunction in cancer. Nat Rev Cancer 2019;19:326-38. [Crossref] [PubMed]
- Johnson J, Thijssen B, McDermott U, et al. Targeting the RB-E2F pathway in breast cancer. Oncogene 2016;35:4829-35. [Crossref] [PubMed]
- Duronio RJ, Brook A, Dyson N, et al. E2F-induced S phase requires cyclin E. Genes Dev 1996;10:2505-13. [Crossref] [PubMed]
- Ma Y, Yuan J, Huang M, et al. Regulation of the cyclin D3 promoter by E2F1. J Biol Chem 2003;278:16770-6. [Crossref] [PubMed]
- Geng Y, Eaton EN, Picón M, et al. Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene 1996;12:1173-80. [PubMed]
- Jagarlamudi KK, Shaw M. Thymidine kinase 1 as a tumor biomarker: technical advances offer new potential to an old biomarker. Biomark Med 2018;12:1035-48. [Crossref] [PubMed]
- Topolcan O, Holubec L Jr. The role of thymidine kinase in cancer diseases. Expert Opin Med Diagn 2008;2:129-41. [Crossref] [PubMed]
- Broët P, Romain S, Daver A, et al. Thymidine kinase as a proliferative marker: clinical relevance in 1,692 primary breast cancer patients. J Clin Oncol 2001;19:2778-87. [Crossref] [PubMed]
- Romain S, Spyratos F, Descotes F, et al. Prognostic of DNA-synthesizing enzyme activities (thymidine kinase and thymidylate synthase) in 908 T1–T2, N0–N1, M0 breast cancers: A retrospective multicenter study. Int J Cancer 2000;87:860-8. [Crossref] [PubMed]
- Bonechi M, Galardi F, Biagioni C, et al. Plasma thymidine kinase-1 activity predicts outcome in patients with hormone receptor positive and HER2 negative metastatic breast cancer treated with endocrine therapy. Oncotarget 2018;9:16389-99. [Crossref] [PubMed]
- McCartney A, Bonechi M, De Luca F, et al. Plasma Thymidine Kinase Activity as a Biomarker in Patients with Luminal Metastatic Breast Cancer Treated with Palbociclib within the TREnd Trial. Clin Cancer Res 2020;26:2131-9. [Crossref] [PubMed]
- Morris LG, Kaufman AM, Gong Y, et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat Genet 2013;45:253-61. [Crossref] [PubMed]
- Nishikawa Y, Miyazaki T, Nakashiro K, et al. Human FAT1 cadherin controls cell migration and invasion of oral squamous cell carcinoma through the localization of β-catenin. Oncol Rep 2011;26:587-92. [PubMed]
- Martin D, Degese MS, Vitale-Cross L, et al. Assembly and activation of the Hippo signalome by FAT1 tumor suppressor. Nat Commun 2018;9:2372. [PubMed]
- Schoninger SF, Blain SW. The Ongoing Search for Biomarkers of CDK4/6 Inhibitor Responsiveness in Breast Cancer. Mol Cancer Ther 2020;19:3-12. [Crossref] [PubMed]
- Li Z, Razavi P, Li Q, et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018;34:893-905.e8. [Crossref] [PubMed]
- Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, et al. Clinical significance of plasma cell-free DNA mutations in PIK3CA, AKT1, and ESR1 gene according to treatment lines in ER-positive breast cancer. Mol Cancer 2018;17:67. [Crossref] [PubMed]
- Zhang Y, Sun X, Icli B, et al. Emerging Roles for MicroRNAs in Diabetic Microvascular Disease: Novel Targets for Therapy. Endocr Rev 2017;38:145-68. [Crossref] [PubMed]
- Majchrzak A, Witkowska M, Smolewski P. Inhibition of the PI3K/Akt/mTOR signaling pathway in diffuse large B-cell lymphoma: current knowledge and clinical significance. Molecules (Basel, Switzerland) 2014;19:14304-15. [Crossref] [PubMed]
- Jansen VM, Bhola NE, Bauer JA, et al. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer Res 2017;77:2488-99. [Crossref] [PubMed]
- Michaloglou C, Crafter C, Siersbaek R, et al. Combined Inhibition of mTOR and CDK4/6 Is Required for Optimal Blockade of E2F Function and Long-term Growth Inhibition in Estrogen Receptor-positive Breast Cancer. Mol Cancer Ther 2018;17:908-20. [Crossref] [PubMed]
- Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010;10:116-29. [Crossref] [PubMed]
- Zhou Y, Wu C, Lu G, et al. FGF/FGFR signaling pathway involved resistance in various cancer types. J Cancer 2020;11:2000-7. [Crossref] [PubMed]
- Chang J, Liu X, Wang S, et al. Prognostic value of FGFR gene amplification in patients with different types of cancer: a systematic review and meta-analysis. PLoS One 2014;9:e105524 [Crossref] [PubMed]
- Haines E, Chen T, Kommajosyula N, et al. Palbociclib resistance confers dependence on an FGFR-MAP kinase-mTOR-driven pathway in KRAS-mutant non-small cell lung cancer. Oncotarget 2018;9:31572-89. [Crossref] [PubMed]
- Formisano L, Lu Y, Servetto A, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun 2019;10:1373. [Crossref] [PubMed]
- Schmid P, Cortes J, Pusztai L, et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N Engl J Med 2020;382:810-21. [Crossref] [PubMed]
- Teh JLF, Aplin AE. Arrested Developments: CDK4/6 Inhibitor Resistance and Alterations in the Tumor Immune Microenvironment. Clin Cancer Res 2019;25:921-7. [Crossref] [PubMed]
- Cooper AB, Sawai CM, Sicinska E, et al. A unique function for cyclin D3 in early B cell development. Nat Immunol 2006;7:489-97. [Crossref] [PubMed]
- Sicinska E, Aifantis I, Le Cam L, et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell 2003;4:451-61. [Crossref] [PubMed]
- Coppé JP, Desprez PY, Krtolica A, et al. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010;5:99-118. [Crossref] [PubMed]
- Deng J, Wang ES, Jenkins RW, et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov 2018;8:216-33. [Crossref] [PubMed]
Cite this article as: Huang W, Wang H. Potential biomarkers of resistance to CDK4/6 inhibitors: a narrative review of preclinical and clinical studies. Transl Breast Cancer Res 2021;2:12.

